How the ReMap catalogue was constructed ?
Which information can you find on ReMap ?
- Annotation and classification of transcription factors
- Genomic visualization of peaks and analyses
- Datasets quality assessment
- DNA constraints under non-redundant peaks
- Downloading peaks and sequences
How the annotation tool works ?
Credits
Datasets sources
We analysed 2,829 quality controlled ChIP-seq experiments from ENCODE and public sources (GEO, ArrayExpress). The public ChIP-seq datasets (n=1,763) as well as the ENCODE ChIP-seq data (n=1,066) have been mapped to the GRCh38/hg38 human assembly. Here we define a “dataset” as a ChIP-seq experiment in a given series (e.g. GSE46237), for a given TF (e.g. NR2C2), in a particular biological condition (i.e. cell line, tissue type, disease state or experimental conditions ; e.g. HELA). Datasets were labeled by concatenating these three pieces of information such as GSE46237.NR2C2.HELA.
For each transcription factor and each cell type, two tables recapitulate the datasets used. A green table represents the datasets used to create the catalogue of binding sites and a red table the datasets filtered out after applying quality filters.
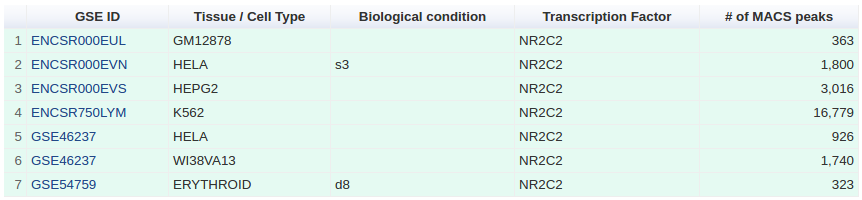

Integration of Public and ENCODE data
ENCODE and Public data have been analysed to propose an unified integration of both data sources, producing a unique atlas of regulatory regions for 485 TRs. We found 125 TRs common to the two sets, 154 proteins specific to ENCODE and 206 specific to the Public catalogue. Taken separately, the ENCODE peaks overlaps by 96% the Public regions, and 87% of the Public peaks overlap ENCODE regions
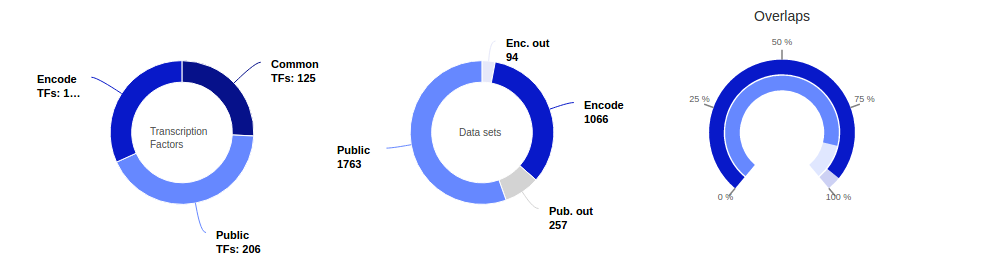
Catalogues of All peaks and Non-redundant peaks
After consistent peak calling, we identified 49 million peaks bound by transcription factors (80 million with ENCODE data included). These numbers include overlapping sites for identical TRs which were studied in various conditions. To address this we merged overlapping TF binding sites for similar TFs obtaining a catalogue of 23.7 million non-redundant binding sites (35.5 million with ENCODE data).
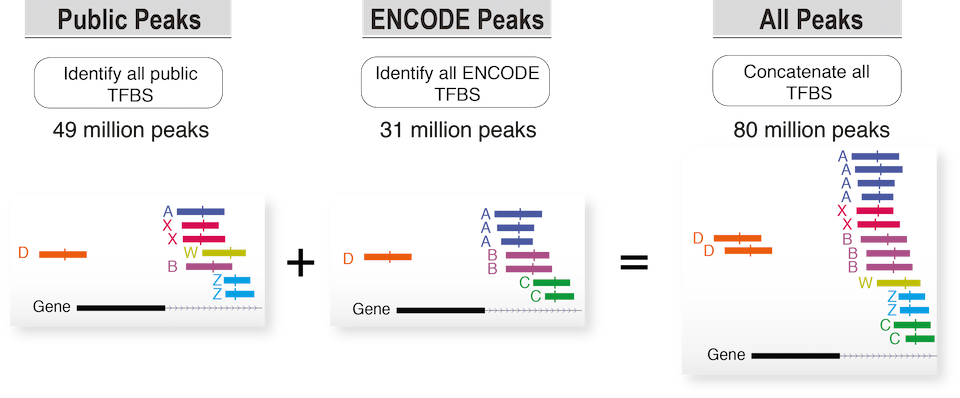
Annotation and classification of transcription factors
Function and description of transcription factors present in this catalogue (Public, Public+Encode) were retrieved from HGNC and RefSeq databases. Each transcription factor was also annotated using the classification of human transcription factors allowing users to filter specific TFs based on the characteristics of their DNA-binding domains.
Genomic visualization of peaks and analyses
To perform a de novo motifs analysis for each TF present in our catalogue, we provide a link to the Regulatory Sequence Analysis Tools. A link to the UCSC Genome Browser was also added to facilitate genomic integration of the binding sites with other genome annotations. Our BED tracks allow for the visualization of our catalogues of binding sites on the human genome. Finally, different analyses such as the quality of datasets and DNA constraint analysis are provided for each transcription factor.

Datasets quality assessment
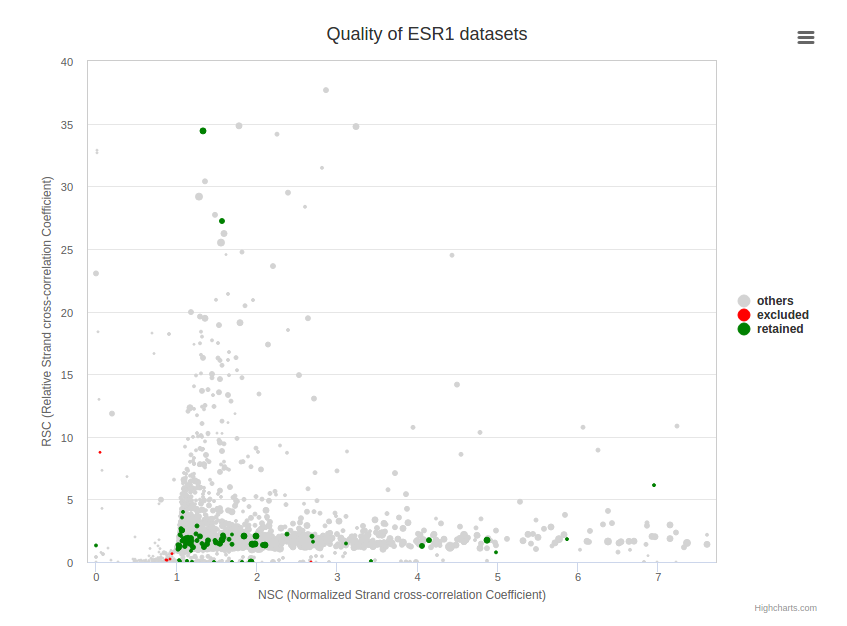
As not every ChIP-seq datasets are equal in terms of quality, we used four different metrics based on ENCODE ChIP-seq guidelines to retain high quality datasets for downstream analyses. First we used the normalized strand cross-correlation coefficient (NSC) which is a normalized ratio between the fragment-length cross-correlation peak and the background cross-correlation, and the relative strand cross-correlation coefficient (RSC), a ratio between the fragment-length peak and the read-length peak to exclude low quality datasets. We also used the fraction of reads in peaks (FRiP) and the number of peaks identified in each dataset to filter datasets.
A bubble plot is available on each page representing the quality of selected dataset(s). Dataset(s) are plotted in a 2D vizualization with NSC and RSC as x- and y-axis, the size of points is correlated with the number of peaks identified in the dataset and colours highlight the datasets conserved (green) or excluded (red) from the catalogue of binding sites.
DNA constraints under non-redundant peaks
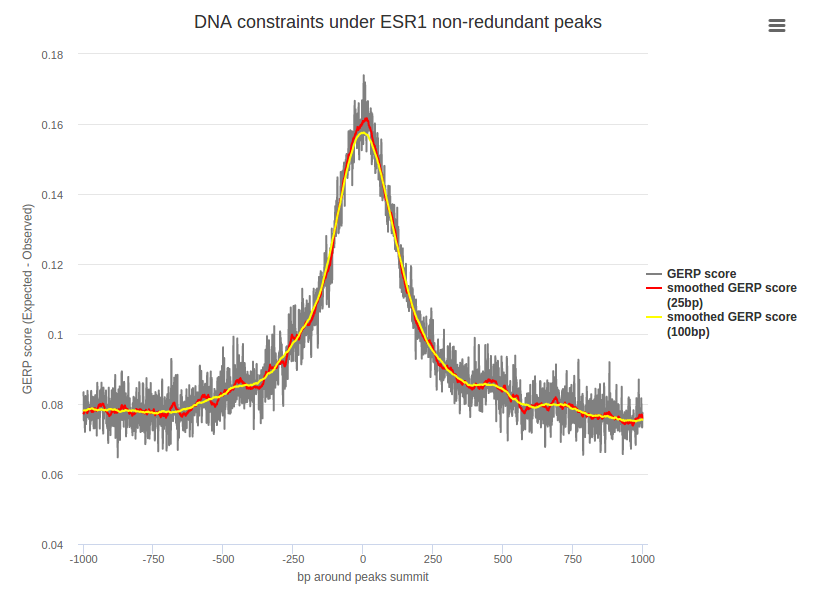
DNA conservation around transcription factor binding sites was computed using the Genomic Evolutionary Rate Profiling (GERP) score obtained and extracted from the Ensembl Compara database for Ensembl v89 using the Compara Perl API. Detailed information can be found at Ensembl and the Sidow Lab.
A plot is available for each transcription factor representing the average DNA constraint for each nucleotide under peak summits.
Downloading peaks and sequences
For each transcription factor, cell type and dataset, we provide files to download peaks in BED format and sequences in FASTA format. The entire catalogue is openly accessible to the community and available to download in BED format. Please do not hesitate to contact us if you need this catalogue in a different format.
Annotation Tool
We provide an annotation tool to annotate user's submitted regions with our catalogue of transcription factor binding sites and to compute statistical enrichments of TFs in these regions.
To do this, we first use intersectBed tool from the BEDTools suite to identify which binding sites of our catalogue fall into the submitted regions. Then, we compare the number of these overlapping regions with the number of overlaps obtained with random regions (same size and number as the submitted regions) to calculate the enrichment of TFs.
Several settings are used by this tool :
- the first option allows you to extend genes (in upstream and/or downstream) before processing the overlap of our catalogue with regions defined around the genes submitted.
This option is available only if a list of Ensembl IDs is provided. - the two next options indicate the minimum percentage of overlap required, and on which data you want to apply this percentage (submitted data or our catalogue). By default, each of our binding sites must overlap your regions by at least 10% to be take into account.
- the last option indicate if the minimum percentage of overlap needs to be applied on both data (submitted data and our catalogue).
Highcharts
This site uses the Highcharts library following the Highcharts licence under the Creative Commons Attribution-NonCommercial 3.0 License (CC BY-NC) as a non-profit / University site (Aix-Marseille University).